
章节实例:第6章 染色质免疫共沉淀测序数据分析
第一部分:峰的寻找(peak calling)
ChIP-seq数据处理最关键的一步是peak ,calling,这里选择FindPeaks软件为例介绍相关的分析方法。FindPeaks被包含在一个简称为Vancouvershortr的软件包里,目前的版本是4.0.16,要求java版本1.6或1.6以上,下载地址:http://sourceforge.net/projects/vancouvershortr/。
该软件支持多种输入格式,包括BED、Bowtie、Eland、Eland Export、GFF、MapView、Map、BAM/SAM。这里以BED格式的输入文件为例概述linux系统环境下软件操作流程。该例中的数据是转录因子FoxA1的ChIP-seq数据,包括Input_tags.bed和对照组数据Treatment_tags.bed。
首先把下载的VancouverShortR-4.0.16.tar.gz包解压,然后在命令行窗口中找到解压后的文件夹并找到其中的一个名为fp4的文件夹。
FindPeaks处理BED格式的文件分为三步:
第一步:先把一个完整的BED文件按染色体号分成数个文件。
1)处理Input_tags.bed,命令如下:
$java –Xmx2G –jar SeparateReads.jar bed /home/starlet/FoxA1/Input_tags.bed /home/starlet/FoxA1/input/
其中,-Xmx2G表示设定的java运行的虚拟机内存为2G;bed表示输入文件格式为bed;/home/starlet/FoxA1/Input_tags.bed为输入文件所在的路径;/home/starlet/FoxA1/input/表示输出文件放在该路径下一个名为input的文件夹中。
2)处理Treatment_tags.bed,命令如下:
$java –Xmx2G –jar SeparateReads.jar bed /home/starlet/FoxA1/Treatment_tags.bed /home/starlet/FoxA1/treatment/
第二步:把第一步中分好的文件中的小片段reads按在基因组中的位置排序。
1)处理Input_tags.bed,命令如下:
$java –Xmx2G –jar SortFiles.jar bed /home/starlet/FoxA1/input/ /home/starlet/FoxA1/input/*.part.bed.gz
其中,/home/starlet/FoxA1/input/表示输出文件存放的路径;/home/starlet/FoxA1/input/*.part.bed.gz表示输入文件所在的路径,其中的*号代表所有染色体号,即代表了上述第一步所生成的结果文件。
2)处理Treatment_tags.bed,命令如下:
$java –Xmx2G –jar SortFiles.jar bed /home/starlet/FoxA1/treatment/ /home/starlet/FoxA1/treatment/*.part.bed.gz
第三步:寻找peaks的步骤,命令如下:
$java –Xmx2G –jar FindPeaks.jar –input /home/starlet/FoxA1/input/*.part.bed.gz –control /home/starlet/FoxA1/treatment/*.part.bed.gz –alpha 0.05 –control_type 0 –aligner bed –output /home/starlet/FoxA1/ -dist_type 1 –name sample –one_per
其中,input /home/starlet/FoxA1/input/*.part.bed.gz表示输入文件所在路径,即步骤1、3所得到的结果文件;–control /home/starlet/FoxA1/treatment/*.part.bed.gz表示输入的对照组文件所在路径,是步骤2、4所得到的结果文件;–alpha 0.05是关于置信区间的参数,值必须在0到1之间,默认的值为0.05,在程序运算中的预测置信区间等于(1-alpha)*100; –control_type 0是在4.0.9.1版本才出现的新参数,它提供两种选择,参数值为0和1,0代表“comparison”基本方法,1代表还在测试中的“hyperbolic section”方法,默认值为0; –aligner bed表示处理的文件格式为BED;–output /home/starlet/FoxA1/表示该步骤输出文件所在的路径; -dist_type 1表示该软件算法中采用的一种片段长度分布模型,1表示triangle distribution,参数值还可以选0,2,3,分别表示fixed width model,Adaptive (sampled) distribution,Native mode, 不过2所代表的Adaptive (sampled) distribution目前不支持,程序默认和推荐的参数为1;–name sample用来给该过程的命名,即过程名为sample;–one_per表示输出的结果文件也根据染色体号分为多个文件, 如果不加这个参数,所有染色体上对应的结果会被放在一个文件内。
如果没有对照组文件,在命令行中去掉-control,-alpha,-control_type三个参数即可运行。
完成上述操作步骤后找到的所有peaks都被存在第三步中输出文件路径下的一个名为sample_triangle_standard.
peaks的文件内,是以参数过程名和所选的算法模型的类型命名的;结果文件还包括每个染色体对应的wig.gz文件以及其它信息文件;还有sample.log文件,以过程名命名的包含程序运行信息的文件。
第二部分:基因关联(gene associate)
PeakAnalyzer的功能是了解peaks处在基因组序列中的位置及其附近区域所包含的基因或者其它诸如启动子等生物学元件。该软件用java实现,下载地址:http://www.bioinformatics.org/peakanalyzer/wiki/Main/Download。由于具有用户界面,所以只要下载解压后找到文件夹中的PeakAnalyzer.jar文件双击即可运行(注意连接网络)。 或者在命令行中输入:
java -jar PeakAnalyzer.jar
运行界面如下:
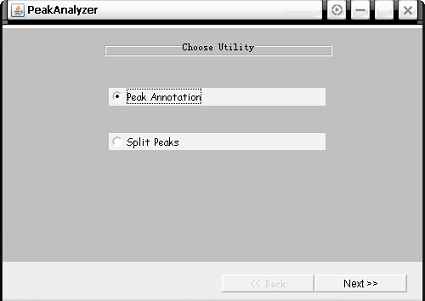
图6-1 PeakAnalyzer的运行界面
首先出现的窗口会让你选择“Peak Annotation”和“Split Peaks”,选择前者然后点“Next”。之后出现的窗口有三个选择“NDG-Nearest Downstream Genes”,“TSS-Nearest Transcription Start Site”和“ODS-Overlapping Data Sets(peak files)”,我们为了找到峰最近的下游基因和与峰区域重合的基因,我们选择第一项NDG点“Next”。
在接下来的窗口中,就可以导入之前用软件找到的峰的文件,注意要把文件中包含“chrom start end”这些列名信息的头一行去掉;然后选择合适的注释文件“Annotations file”,选择“Coding genes only”或“Coding and non-coding
genes”,设置好“symbol file”(当注释文件为GTF格式时不需要),输出文件路径和“Prefix”(这里指输出文件名)。都设置好后点“Next”程序即运行,运行完后还会让你选择是否生成pdf的图,如果你装了统计软件R,可以选择生成,当然结果还包括了包含下游或重合基因信息的BED格式的文件。
第三部分:Motif发现(motif discovery)
打开浏览器,输入MEME的网址:http://meme.sdsc.edu/meme4_5_0/cgi-bin/meme.cgi,按照页面上的要求输入你的邮箱地址,你得到的peaks的序列fasta文件,设定motif的宽度和数量等信息,完成之后点击“Start search”。等待一段时间后,它会帮你找出motif,并把结果的链接发到你的邮箱。为了了解MEME找出的motif是否与已知的motif相似,可以用TOMTOM。在MEME的结果网页上的motif图案的下方会有“TOMTOM”的按钮,点击就会将该motif和已知的数据库中的motif进行比对。TOMTOM的motif数据库包括JASPAR,TRANSFAC和UNIPROBE,网址:http://meme.sdsc.edu/meme4_5_0/cgi-bin/tomtom.cgi,可直接在该页面上输入motif就可进行分析。

图6-2 MEME提交数据的页面
第四部分:注释分析(GO,KEGG analysis)
线生物学分析工具DAVID可以完成GO和KEGG的pathway富集分析。网址:http://david.abcc.ncifcrf.gov/。点击菜单栏的“Start Analysis”按钮,进入分析界面。
第一步:在提交窗口中导入之前所得到的峰附近的基因列表,选择“gene list”单选框,单击上传“upload”按钮;
第二步:选择物种,在上传完成的窗口中选择“human”作为分析的物种,单击“use species”按钮;
第三步:在服务器自动选择人类所有基因作为背景的前提下,选择要富集的层次,点击富集按钮,将弹出所选择层次上的富集结果。同时,可以单击列表右上方的“download”超链接下载结果文件。
第五部分:可视化(visualization)
UCSC的genome browser是比较常用的对序列进行可视化的工具,网址:http://genome.ucsc.edu/。进入主页后点击窗口左边竖列的第一项“Genome Browser”,就会进入Genome Browser Gateway。选择合适的物种基因组后点击页面中央的“add custom tracks”按钮即可进入数据上传页面,用户可以直接贴上数据或者数据的链接地址,也可以点击“选择文件”按钮以导入你之前的ChIP-seq的数据,选好后点击“submit”按钮即可提交。等待一段时间,在数据上传成功后的页面上点击右侧的“go to genome browser”按钮即进入结果页面。
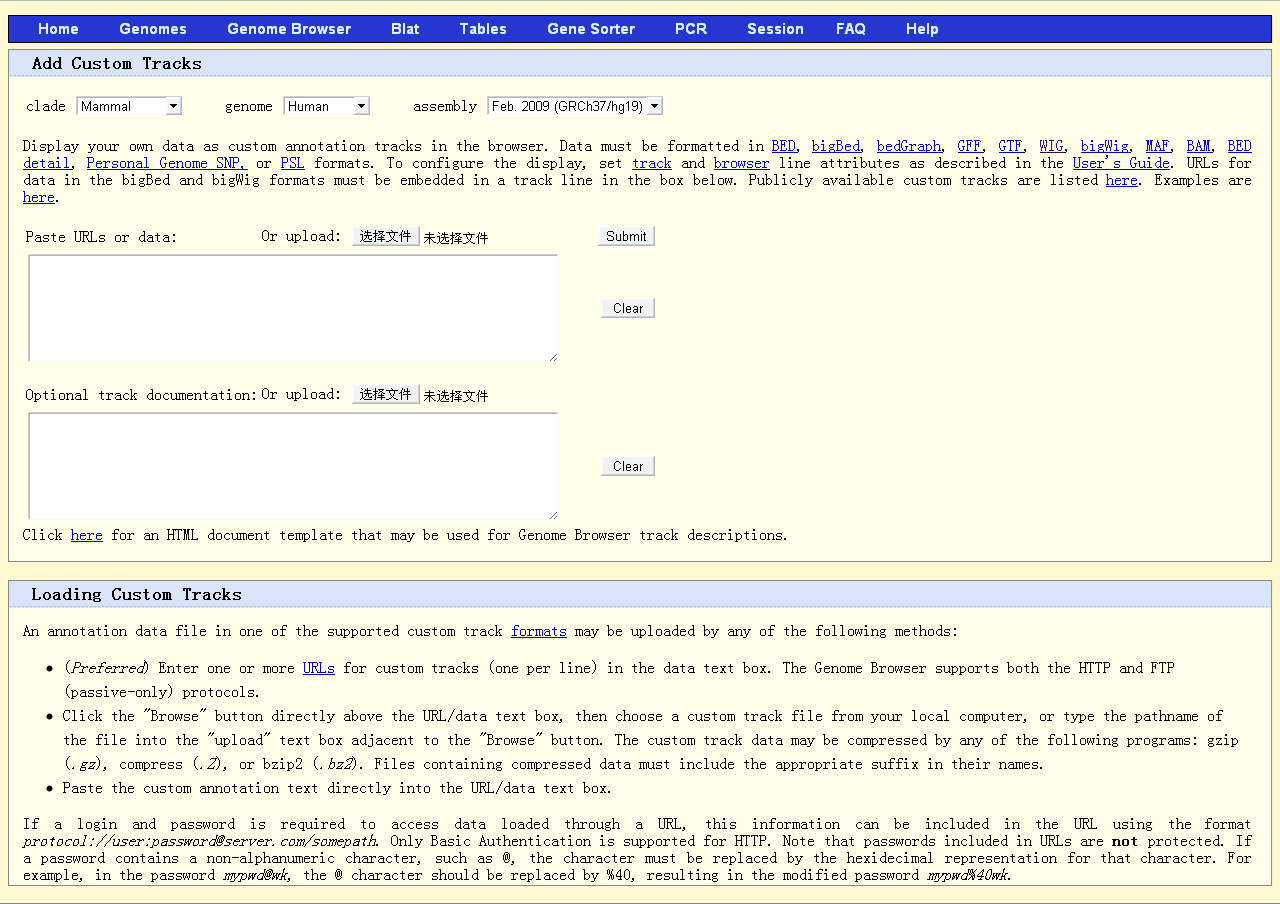
图6-3 CSC上提交数据的页面
关于我们

苏州大学系统生物学研究中心

苏州大学