
章节实例:第8章 MicroRNA-Seq数据分析
第一部分:本地分析软件
(一) mireap
mireap下载地址为:https://sourceforge.net/projects/mireap/,运行环境为支持Perl语言的Linux或Windows操作系统。软件输入文件为FASTA格式,输出有三个文件,分别以*.gff、*.aln和*.log后缀结尾。
软件安装步骤如下:
1)安装ViennaRNA package 2.0,下载地址为:http://www.tbi.univie.ac.at/RNA/。请务必确保计算机可以正常运行Perl脚本程序;
2)将下载完成的mireap_0.2.tar.gz文件拷贝至目录/foo/bar,使用如下命令解压缩该文件;
tar –zxvf
3)运行mireap之前,需要增加一个可以运行Perl脚本程序的路径。其中,csh/tcsh用户使用如下命令添加:
setenv PERL5LIB /foo/bar/mireap_0.2/lib
如果是sh/ksh/bash用户,请使用下面这条命令添加:
export PERL5LIB=/foo/bar/mireap_0.2/lib
软件的输入文件包括三个部分,分别是:
1)smrna.fa:miRNA文库文件。将非FASTA格式的miRNA文库文件转换成FASTA格式,并且给每个序列增加一个标号(ID),条目如下:
>t0000035 3234
GAATGGATAAGGATTAGCGATGATACA
>t0000072 1909
TTGCAGTATGTAGGAAATCAAAACGTTC
…(下略)
以第一行为例,t0000035表示序列读段(read)的read_ID,3234表示相应的测序频数(sequencing frequence)。
2)map.txt:miRNA在染色体上的映射(mapping)关系文件,格式如下:
t0000035 nscaf1690 4798998 4799024 +
t0000035 nscaf1690 4805385 4805411 +
t0000035 nscaf1690 7588502 7588528 +
t0000072 nscaf1690 2923961 2923988 -
…(下略)
该文件每一行的相邻元素用Tab键分隔。其中,第一列表示序列读段的read_ID,第二列表示染色体ID,第三列和第四列分别表示读段在染色体上的起始位置和终止位置,第五列的正负号用于区分读段在染色体上的正义链和负义链。
3)ref.fa:参考(reference)序列文件,FASTA格式,内容如下:
>nscaf1690 /length=7983491 /lengthwogaps=7414637
AAAAAAAAAGGACAGTATAAATAGAATAGTAACAAAGGTGAAATTAATTGTTAGTTCGGTTTGTTATGGAAGTATTTTTTTTT
GTTTATAAGCAAATTTGTTTGTTTTTAAA
…(下略)
基于上述三个文件,就可以安装运行软件了。命令如下:
mireap.pl -i <smrna.fa> -m <map.txt> -r <reference.fa> -o <outdir>
其中,相关参数的含义如下:
-i:miRNA文库,FASTA格式
-m:miRNA在染色体上的映射(mapping)关系
-r:参考序列,FASTA格式
-o:结果输出路径(默认:当前文件夹)
软件运行完成后,将输出三个结果文件,分别是:
1)*.gff文件:该文件包含mireap发现的miRNA基因,数据格式为gff3。其中,“Count”表示测序频数;
2)*.aln文件:该文件包含序列、miRNA前体结构和miRNA比对至前体序列的信息;
3)*.log文件:该文件为日志文件,包含相关参数、软件运行的开始和结束时间等附加信息。
(二) miRDeep
miRDeep下载地址:https://www.mdc-berlin.de/8551903/en/research/research_teams/systems_biology_of_
gene_regulatory_elements/projects/miRDeep。网站中提供了软件运行的脚本文件miRDeep.tgz以及测试数据demo_limited.tgz、demo_full.tgz。除此之外,软件运行还需要以下必要文件:
1)NCBI包,下载地址:http://www.ncbi.nlm.nih.gov/Ftp/,其中包含了所有可执行软件和相关文件;
2)Vienna包,下载地址:http://www.tbi.univie.ac.at/RNA/,其中包含了详细的UNIX和Windows用户的安装信息。如果是UNIX用户,软件下载后输入以下编译命令:
./configure
make
sudo make install
下面以深度测序结果454_total.fa和线虫基因组为例,演示软件的使用方法(数据文件详见demo_limited.tgz):
formatdb -i genome_cel.fa -p F -o T
megablast -d genome_cel.fa -i 454_total.fa -o blastout -W 12 -D 2 -p 100
blastoutparse.pl blastout > blastparsed
filter_alignments.pl blastparsed -c 5 > blastparsed_excision
overlap.pl blastparsed_excision ucsc_ncRNA -b > ids_overlap_ucsc_ncRNA
cat ids_overlap_* | sort -u > ids_overlap_total
blastparselect.pl blastparsed_excision -g ids_overlap_total > blastparsed_excision_filtered
filter_alignments.pl blastparsed_excision_filtered -b 454_total.fa > 454_aligned.fa
excise_candidate.pl genome_cel.fa blastparsed_excision_filtered > precursors.fa
cat precursors.fa | RNAfold -noPS > structures
auto_blast.pl 454_aligned.fa precursors.fa -b > signatures
miRDeep.pl signatures structures -s mature_metazoan_no_cel_v10.fa -y > predictions
软件的输出文件名为Predictions,输出数据中包括预测分值、miRNA成熟体/前体/初级体的序列、结构等信息。
(三) miRExpress
miRExpress下载地址:http://mirexpress.mbc.nctu.edu.tw/。该软件编译环境为32或64位的Linux操作系统,并且处理器要求支持SSE3,安装命令如下:
./configure
make
sudo make install
miRExpress适用于FASTQ格式的miRNA深度测序数据处理,序列的碱基数要求小于64。该软件已知的miRNA信息来源于miRBase。原始数据处理以及miRNA表达谱构建的命令流程概括如下:
Raw_data_parse -> Trim_adaptor -> alignmentSIMD -> analysis
具体操作步骤为:
1)使用“Raw_data_parse”命令处理FASTQ格式的原始数据,相应的输出结果是单一的(unique)读段序列以及它们的数量,Tab键分隔,命令格式如下:
Raw_data_parse [-i raw_data] [-o output file name, optional]
其中,相关参数的含义如下:
-i:原始测序数据,FASTQ格式
-o:输出文件名(默认:输入文件名+“.merge”)
2)使用“Trim_adapter”命令处理包含接头(adaptor)的读段序列,输入数据中序列数目和序列之间使用Tab键分隔,格式如下:
命令格式如下:
Trim_adapter [-i input file] [-t 3' adaptor sequence file] [-h 5' adaptor sequence file, optional] [-o output file name, optional]
其中,相关参数的含义如下:
-i:输入的序列文件
-t:3’端接头序列文件
-h:5‘端接头序列文件
-o:输出文件名(默认:输入文件名+“.trim”)
3)使用“alignmentSIMD”命令比对查询序列和参考序列。用法如下:
alignmentSIMD [-r precursor miRNA file] [-i input sequence file] [-o output directory] [-t alignment identity, optional] [-n Rank nohit file] [-u Number of CPU for calculation]
其中,相关参数的含义如下:
-r:miRNA前体
-i:输入文件
-o:输出目录
-t:查询序列和参考序列间的比对增幅(默认值:1)
-n:结果排序文件,按读段序列的数量排序
-u:希望创建的线程数,取决于计算机CPU数量(默认值:1)
4)使用“analysis”命令分析比对结果并且构建miRNA表达谱。用法如下:
analysis [-r precursor miRNA file] [-d alignment result directory] [-o output file name (expression)] [-t output file for comparing result,optional]
其中,相关参数的含义如下:
-r:miRNA前体(与“alignmentSIMD”中使用的文件一致)
-d:比对结果目录(与“alignmentSIMD”中定义的目录一致)
-o:输出文件:比对结果
-t:输出文件:表达谱构建结果
此外,软件提供了相关的测试数据,由于篇幅所限,详细的操作说明请参阅软件的“Example”和“README”文件。
(四) miRTRAP
miRTRAP下载地址:http://flybuzz.berkeley.edu/miRTRAP.html。使用如下命令进行安装:
gunzip miRTRAP.tar.gz
tar xvf miRTRAP.tar
cd miRTRAP
perl makefile.pl
make
make test
make install
软件的输入文件主要有两个:1)配置文件:config.txt;2)读段信息文件:reads.txt。
可以使用如下命令运行软件:
./miRTRAP.pl config.txt
此外,软件也可以分步运行。由于篇幅所限,详细的步骤方法请参阅相关的测试数据文件。
(五) MIReNA
MIReNA下载地址:http://www.ihes.fr/˜carbone/data8/,以深度测序数据处理为例,软件安装运行命令如下:
1)软件安装:
cd <repository where this README is>
./configure
make
2)软件运行:
../MIReNA.sh -D -b cel_deep_sequencing/megablastparsed_filtered -f cel_deep_sequencing/454_total.fa -j cel_deep_sequencing/genome_cel.fa -k cel_deep_sequencing/mature_metazoan_no_cel_v14.fa
这里,454_total.fa是深度测序得到的结果文件,FASTA格式;genome_cel.fa是参考基因组信息,FASTA格式。由于篇幅所限,详细内容请参阅相关的测试数据文件。
(六) miRNAkey
miRkey下载地址:http://ibis.tau.ac.il/miRNAkey/,网站中提供了软件的可执行文件、测试数据以及相应的操作说明。软件的运行环境为64位架构的Linux/Unix 或Mac系统,除此之外,需要手动安装Java运行环境(JRE,6.0版本及以上)、Burrows-Wheeler Alignment工具、Fastx-Toolkit以及以下Perl模块:
Math::CDF
Spreadsheet::WriteExcel
Getopt::Long
GD::Graph::bars
以上工具可以点击http://ibis.tau.ac.il/miRNAkey/req.html选择下载,Perl模块可以通过如下命令安装:
$>cpan Math::CDF Spreadsheet::WriteExcel Getopt::Long GD::Graph::bars
下载完成后,可以通过双击miRNAkey.jar文件运行软件,以网站提供的测试数据“Small sample-data & output”为例,软件操作如图8-1所示。
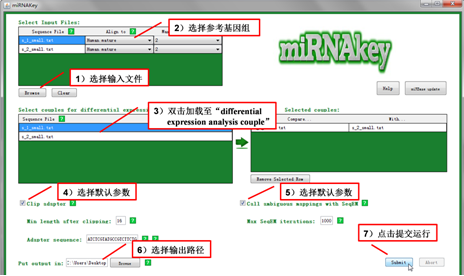
图8-1 miRNAkey软件操作示意图
软件的输出文件包括比对结果、剪切结果、比对上和未比对上的读段序列、SEQEM分析结果、差异表达分析结果、总结报告等。详细信息请参阅测试数据 “sample_output_data”文件夹中的相关内容。
第二部分:在线分析软件
(一) miRanalyzer
miRanalyser的网址为:http://web.bioinformatics.cicbiogune.es/microRNA/。该软件允许的输入数据格式有两种:
1)包含读段序列和实验中该读段出现次数的文本文件,其中,读段序列和次数之间使用Tab键分隔,格式如下:
GAGGTAGTAGGTTGTA 49862
ACCCGTAGAACCGACC 15490
…(下略)
2)FASTA格式,参考如下:
>ID 49862
GAGGTAGTAGGTTGTA
>ID 15490
ACCCGTAGAACCGACC
…(下略)
其中,“49862”表示实验中读段“GAGGTAGTAGGTTGTA”出现的次数。详细信息请参阅:http://web.bioinformatics.
cicbiogune.es/microRNA/manual.html。
文件上传后需要设置必要的运行参数,如图8-2所示。
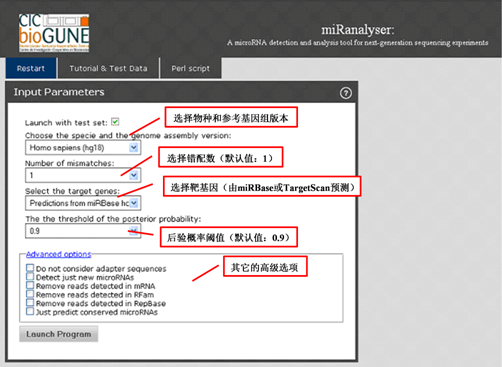
图8-2 miRanalyzer运行参数设置示意图
软件的运行结果主要包括:1)输入数据的总结报告;2)输入数据中包含已知miRNA的统计结果;3)与其它转录组序列比对的统计结果;4)预测到的新miRNA的统计结果;5)未匹配的读段信息等。
(二) DSAP
DSAP的网址为:http://dsap.cgu.edu.tw,输入数据的格式如下:
260135 TGGAATGTAAAGAAGTATGTATTCGTATGCCGT
213816 TGAGGTAGTAGGTTGTATAGTTTCGTATGCCGT
…(下略)
其中,第一列表示读段序列的拷贝数,第二列表示读段序列,使用Tab键分隔。如果文件为FASTQ格式,可以从网站http://maasha.github.io/biopieces/下载工具read_fastq将格式转换成要求的输入形式,命令如下:
read_fastq -i INPUT.fastq | uniq_seq -c | sort_records -r -k SEQ_COUNTn | write_tab -k SEQ_COUNT,SEQ -xo OUTPUT.tag
上传完成后,设置必要的运行参数,例如,是否考虑接头序列、是否考虑多聚A/T/C/G/N核苷酸、选择物种等。软件运行结果通过图表等形式直观呈现,如图8-3所示。
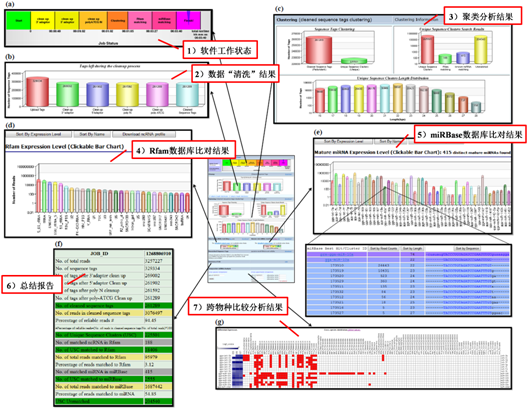
图8-3 DSAP运行结果汇总
(三) mirTools
mirTools的网址为:http://centre.bioinformatics.zj.cn/mirtools/,软件输入为原始测序数据经过预处理后的FASTA文件,格式如下:
>UniTag-009_x80
CATTTATTATTTATCTTATTCCTTCTTCTTTTTTA
…(下略)
这里,“UniTag-009”表示读段序列的唯一ID,由用户定义;“x80”表示“UniTag-009”标记的读段序列在测序样本中出现了80次,它们之间使用下划线“_”相连。网站http://centre.bioinformatics.zj.cn/mirtools/adaptortrim.php提供了Perl脚本程序Adapter_trim,用于从原始测序数据中去除低质量的读段序列以及3’/5’接头序列。使用命令如下:
perl Adapter_trim.pl [options] >outputfile
其中,相关参数的含义如下:
-i:FASTQ格式的短序列文件
-n:样本名称(默认:sample)
-x:5’端接头序列(默认:GTTCAGAGTTCTACAGTCCGACGATC)
-y:3’端接头序列(默认:TCGTATGCCGTCTTCTGCTTG)
-f:FASTQ文件格式,1:Sanger格式;2:Solexa/Illumina 1.0格式;3:Illumina 1.3+格式。(默认值:2)
软件运行输出包括已知miRNA比对识别、新的miRNA预测、miRNA差异表达分析等结果,如图8-4所示。相关内容会通过电子邮件反馈给用户。
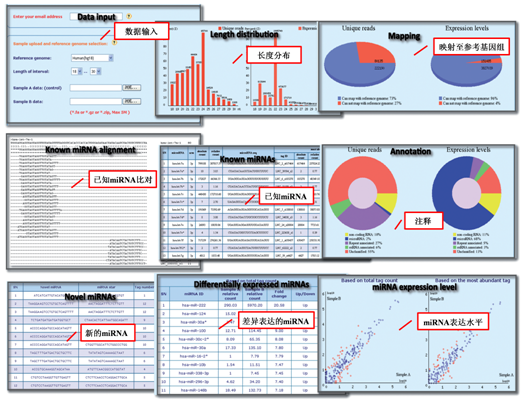
图8-4 mirTools运行结果汇总
关于我们

苏州大学系统生物学研究中心

苏州大学