
章节实例:第7章 转录组测序数据分析
第一部分:选择性剪切软件
(一) Tophat
TopHat是最常用的分析选择性剪接的工具,这里选择的版本为2.0.9,Linux版。操作系统选择Ubuntu(v10.04)。在安装TopHat之前,首先需要在系统中安装samtools工具和Boost C++库。 这里,samtools版本是0.1.19,下载地址:http://samtools.sourceforge.net/,Boost版本是1.54.0,下载地址:http://www.boost.org/users/download/。
在TopHat的官方网站中,提供了各种版本的TopHat软件,其中包括已编译完成和未编译过的文件。用户可以从http://tophat.cbcb.umd.edu/downloads/选择下载。这里,我们将介绍TopHat编译和安装的过程。为了简单起见,默认的安装路径均为/usr/local, 用户可以根据实际情况在Linux环境中通过--prefix命令选择合适的安装路径。相应的方法也可以参考TopHat网站中的内容,网站地址:http://tophat.cbcb.umd.edu/tutorial.shtml。
需要注意的是,在安装samtools、Boost和TopHat过程中,首先需要保证系统中存在必要的依赖文件,这些文件可以通过以下命令下载安装:
1)安装samtools需要ncurses:
$ sudo apt-get install libncurses5-dev
2)解决samtools安装过程中“zlib.h:没有那个文件或目录”的问题:
$ sudo apt-get install zlib1g-dev
3)解决boost编译过程中出现的问题:
$ sudo apt-get install python-dev gccxml
4)解决tophat中configure过程中的编译问题:
$ sudo apt-get install g++
确保上述依赖文件存在后,首先,编译samtools。下载软件并转到软件所在的当前目录,根据以下命令进行编译:
$ bunzip2 samtools-0.1.19.tar.bz2
$ tar xvf samtools-0.1.19.tar
$ cd samtools-0.1.19
$ make
编译完成后,复制samtools文件到/usr/local/bin目录下:
$ cp -a samtools /usr/local/bin
根据TopHat说明文件要求,需要在/usr/local/include/下建立一个bam目录:
$ mkdir /usr/local/include/bam
切换到samtools目录下将samtools目录下的所有.h文件拷贝至/usr/local/include/bam/下:
$ cp -a *.h /usr/local/include/bam
将libbam.a文件拷贝至/usr/local/lib/目录下:
$ cp -a libbam.a /usr/local/lib
这样就完成了samtools的编译和安装。
接下来对Boost进行编译:第一步,解压并编译bjam:
$ tar zxvf boost_1_54_0.tar.bz2
$ cd boost_1_54_0
$ ./bootstrap.sh
第二步,编译Boost:
$ ./bjam link=static \runtime-link=static stage install
第三步,更新动态链接库:
$ sudo ldconfig
正确编译和安装samtools和Boost后就可以进入TopHat的编译安装过程了:
$ tar zxvf tophat-2.0.9.tar.gz
$ cd tophat-2.0.9
$./configure –with-boost=/usr/local \--with-bam=/usr/local
$ make
$ make install
这样就完成了TopHat的编译和安装过程,其可执行文件在”/user/local/bin/”目录中。由于Tophat功能的实现依赖软件Bowtie,所以必须将Bowtie,Bowtie-build和Bowtie-inspect这三个可执行文件复制到”/user/local/bin/”目录中。
$ cp –a bowtie bowtie-build bowtie-inspect /usr/local/bin
Tophat的使用。首先,可以从地址http://tophat.cbcb.umd.edu/downloads/test_data.tar.gz中下载用于测试软件安装结果的文件test_data.tar.gz,转到该文件所在目录,解压文件并使用tophat命令测试安装结果:
$ tar zxvf test_data.tar.gz
$ cd test_data
$ tophat –r 20 test_ref reads_1.fq reads_2.fq
输出文件位于当前目录的“tophat_output”文件夹下,其中有两个文件“accepted_hits.bam”和”junctions.bed”以及一个名为“logs”的文件夹,该文件夹下存储了命令行、参数设置等运行内容。“accepted_hits.bam”为bam格式的二进制比对结果文件,无法使用普通的文本编辑工具打开,可以使用已经安装的samtools工具转换成sam格式的文件后查看内容,命令行为:
$ cd tophat_output
$ samtools view –o accepted_hits.sam accepted_hits.bam
转换后可以用文本剪辑工具打开文件。命令行输入”more accepted_hits.sam”或”gedit accepted_hits.sam”。这里,每一条记录为一条序列片段与参考基因组的比对结果,如图7-1所示。具体参考格式说明可以参考网页说明http://genome.ucsc.edu/FAQ/FAQformat.html,另一个文件”junctions.bed”为用bed格式存储的预测到的可变剪接位点信息,如图7-2所示。
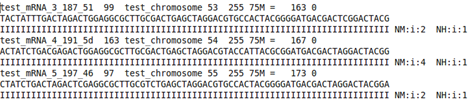
图7-1 序列片段与参考基因组的比对结果示例
![]()
图7-2 预测到的可变剪接位点信息示例
(二) HMMSplicer
HMMSplicer的下载地址为:http://derisilab.ucsf.edu/software/HMMSplicer/hmmSplicer-0.9.5.tar.gz,当前选择版本是0.9.5,Linux版。下载得到的HMMSplicer使用Python语言编写,可以不需要编译直接运行。但是,其运行需要依赖Python2.6(http://python.org)、numpy和Bowtie。
首先,下载并安装Python,由于大多数Linux发行版都自带Python,故其安装在这里不做赘述。使用下面命令安装numpy:
$sudo apt-get install python-numpy
其次,解压下载到的HMMSplicer压缩包,并手动修改Bowtie路径:
$tar xzf hmmSplicer-0.9.5.tar.gz
$cd hmmSplicer-0.9.5
修改Bowtie路径一般有两种方法,一是通过修改HMMSplicer运行配置文件”configVals.py”;二是将Bowtie的路径加到环境变量”PATH”中。下面是两种方法的命令行:
第一种,修改PATH_TO_BOWTIE列为PATH_TO_BOWTIE:
$nano configVals.py ="~/biosoft/bowtie-0.12.7/bowtie"
第二种,修改环境变量,添加Bowtie路径:
$export PATH=$PATH: /home/eragon/biosoft/bowtie-0.12.7/bowtie
设置好以上步骤后,就可以正常使用HMMSplicer了。当然,在使用之前可以先查看帮助文档,熟悉其使用方法,这一步可以通过使用不带参数的运行命令实现:
$python runHMM.py
与其它可变剪接位点预测工具类似,HMMSplicer同样提供了很多的设置参数,包括输出路径、二代测序FASTQ输入文件、FASTA基因组文件等常规参数,以及错配数目、得分阈值、最大及最小内含子长度等默认辅助参数。下面通过简单的实例展示HMMSplicer的运行:
$cd ~/biosoft/hmmSplicer-0.9.5 $python runHMM.py
-o ../hmmsplicer_test/hmmsplicer_output/ -i ../hmmsplicer_test/reads_1.fq
-g ../hmmsplicer_test/test_ref.fa
运行结果如图7-3和图7-4所示。其中,图3显示了Bowtie建立基因组索引及比对结果;图4显示了HMMSplicer预测到的可变剪接位点。

图7-3 Bowtie建立基因组索引及比对结果示例
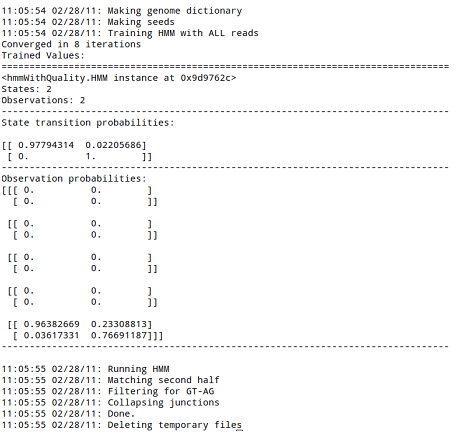
图7-4 HMMSplicer预测到的可变剪接位点示例
第二部分:基因表达水平分析软件
CuffLinks
Cuffinks下载地址为:http://cufflinks.cbcb.umd.edu/downloads,当前选择的版本是2.1.1,操作系统为Ubuntu(v10.04)。CuffLinks的运行也需要samtools和Boost的支持(Boost的版本要求至少为1.47)。 除此之外,还需要Eigen库(这里选择的版本为3.2.0)。对于samtools和Boost的编译和安装方法不再赘述。这里,重点介绍Eigen和Cufflinks的编译安装过程。
与安装TopHat时一致,为了简单起见,默认安装路径仍然为/usr/local(这里要求用户具有root权限),用户可以根据实际情况在Linux环境中通过--prefix命令选择合适的安装路径。相应的方法也可以参考Cufflinks网站中的内容:http://cufflinks.cbcb.umd.edu/tutorial.html。
Eigen库的下载地址为:http://eigen.tuxfamily.org/index.php?title=Main_Page。下载后只需要将文件解压,然后将eigen-3.2.0文件夹中的Eigen子文件夹复制到/user/local/include目录下即可。命令如下:
$ tar zxvf eigen-3.2.0.tar.bz2
$ cd eigen-3.2.0
$ cp –a Eigen /usr/local/include
完成samtools、Boost和Eigen的编译安装后,就可以进行Cufflinks的编译安装了。具体命令如下:
$ tar zxvf cufflinks-2.1.1.tar.gz
$ cd cufflinks-2.1.1
$ ./configure --with-boost=/usr/local/ --with-eigen=/usr/local/ --with-bam=/usr/local/
$ make
$ make install
安装完成后,可以从地址http://cufflinks.cbcb.umd.edu/downloads中下载测试文件test_data.sam,转到该文件所在目录,使用cufflinks命令测试安装结果:
$ cufflinks ./test_data.sam
此外,使用CuffDiff命令可以对两个或以上的样本数据进行表达差异分析,命令行如下:
$ cuffdiff transcripts.gtf sample1.sam sample2.sam
第三部分:综合性分析软件
(一) rQuant.web
rQuant.web将RNA-Seq数据分析过程中需要用到的各种工具进行了有机整合,可视化界面显示,并且提供网络在线服务。最为重要的是,基于二次规划的rQuant技术能够估计来自于文库制备、测序和读段定位过程中的各种偏差,使得分析结果更加准确。
rQuant.web的网站主页地址为:https://galaxy.cbio.mskcc.org/,进入后界面如图7-5所示。网站主页左侧是工具列表。这里可以找到RNA-Seq数据分析过程中用于读段质量检测、数据格式转换、读段定位、选择性剪接事件识别、转录本预测及组装、基因表达分析、富集分析等常用的工具。通过合理选择这些工具,能够很好地处理来自不同测序平台的测序数据。基于一般的RNA-Seq数据分析流程,rQuant.web中整合的工具对应有:
1)读段定位:Bowtie2,BWA,GenomeMapper,BFAST,PerM,Meqablast等;
2)可变剪接比对:TopHat,TopHat2,PALMapper,STAR,ASP等;
3)转录本预测及组装:mTIM,Trinity,Cufflinks,Cuffcompare,Cuffmerge等;
4)基因表达分析:rQuant,rDiff,DESeq,DESeq2,edgeR,Cuffdiff等;
5)富集分析:topGO,GeneSetter等。
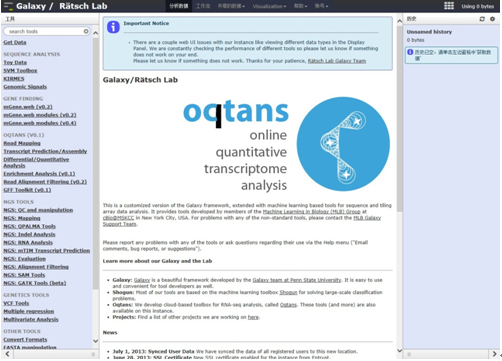
图7-5 rQuant.web网站主页界面
此外,对于读段质量检测,rQuant.web中也整合了如FastQC等工具,对于数据格式转换,rQuant.web能够轻松实现诸如SAM-BAM格式、FASTA-FASTQ格式等的转换。其它类型的功能工具详请参见rQuant.web网站主页。
(二) ArrayExpressHTS (AEHTS)
这里介绍AEHTS基于EBI R-Cloud的用法。EBI R-Cloud是EBI上提供的新服务,可以让R用户们登录并且能够在64位的Linux结点上进行分布式运算的工作。AEHTS具体使用流程如下:
1)下载并安装Java客户端ArrayExpress R/Bioconductor Workbench:http://www.ebi.ac.uk/tools/rcloud。当第一次运行时,需要注册,需要填写用户名、密码以及email等信息。
2)创建一个新项目,如图7-6所示。
图7-6 创建新项目
3)以默认参数运行ArrayExpressHTS。这里以Chepelev et al.等人提供的数据集E-GEOD-16190为例介绍,这个数据集用于研究人类基因组上外显子表达时单核苷酸变异。在控制台输入以下命令:
> library(ArrayExpressHTS)
> e <- ArrayExpressHTS("E-GEOD-16190")
运行后,变量e是一个ExpressionSet对象,便于下游分析。整个流程的运算时间取决于数据的大小和用于计算的结点数。分析报告会在每一步结束后产生。
4)质量报告。随着每一步分析的完成都会产生相应的分析报告。具体到E-GEOD-16190的数据集共会产生如图7-7所示的几个报告:
a. 原始数据E-GEOD-16190一个泳道的质量报告;
b. E-GEOD-16190一个泳道的匹配报告;
c. E-GEOD-16190比较分析报告。

图7-7 基于E-GEOD-16190数据集的分析报告
关于我们

苏州大学系统生物学研究中心

苏州大学